急性胰腺炎(AP)的发生主要经历了胰酶激活、自身消化和炎症反应等过程。关于胰酶激活,国外学者在细胞生物学方面的新认识值得重视。
胰酶激活部位在腺泡细胞
生理情况下,腺泡细胞合成的胰酶经胰管进入肠道,被肠激酶激活后发挥其消化功能。曾经认为AP的启动发生在胰管附近, 但1992年美国哈佛大学消化疾病研究中心在负鼠的AP模型上观察到,结扎胰腺导管3h后,腺泡细胞丧失其极性,腺泡腔扩张;6h后腺泡细胞坏死;12h后小叶坏死、炎症和出血,导管旁没有明显病变。该实验表明,胰酶激活的部位在腺泡细胞,而不在胰管附近。
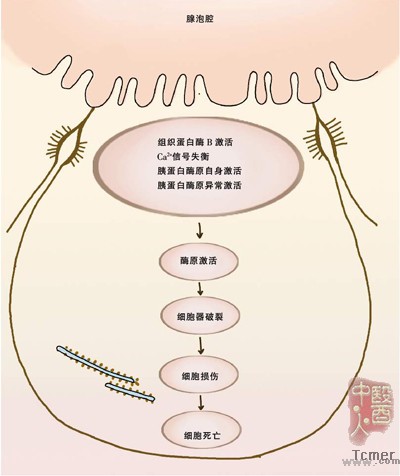
胰分泌液中有许多溶酶体酶,如组织蛋白酶B、芳香基硫化酶等。如果溶酶体酶过早地在腺泡细胞内激活胰蛋白酶原使之成为胰蛋白酶,而胰蛋白酶抑制物含量不足以抵消活化的胰蛋白酶,则将引发一系列胰酶原的活化,导致胰腺的自身消化。
腺泡细胞内消化酶原与溶酶体水解酶相遇是AP早期事件
生理情况下,消化酶原和溶酶体水解酶均在粗面内质网合成,随之被转运到高尔基器。溶酶体水解酶在翻译后先糖基化,然后第6位的甘露糖被磷酸化。高尔基器内有甘露糖-6磷酸化受体,并通过该受体将溶酶体水解酶转运到溶酶体前体内,溶酶体水解酶与甘露糖-6磷酸化受体解离。胰蛋白酶原在粗面内质网-高尔基器-酶原颗粒转运过程中不与甘露糖-6磷酸化受体结合。正是通过这两种不同的途径,同在粗面内质网合成的消化酶原和溶酶体水解酶被最终分选到不同的分泌泡内,分别形成了消化酶原颗粒和溶酶体。腺泡细胞内的溶酶体与细胞顶面膜融合后,有助于消化酶原颗粒的释放。
实际上,不论在何种病因诱导的AP模型中,腺泡细胞对氨基酸的摄取、新蛋白质的合成及从内质网向高尔基器转运等均未发生明显改变。但为溶酶体合成的蛋白酶在细胞器内的分选却发生了显著改变。Saluja 等在多种实验性胰腺炎模型中,采用电镜等技术观察到溶酶体水解酶在胞内的迁移和与消化酶原的分隔现象发生了改变。他们观察到,正常对照组80%的组织蛋白酶B在溶酶体细胞器内,约20%在消化酶原浓缩泡内。但在胰腺炎组,疾病早期,>50%的组织蛋白酶B在消化酶原浓缩泡内,组织蛋白酶 B 含量在溶酶体细胞器内明显降低。免疫荧光定位发现含有消化酶原和溶酶体水解酶的细胞器增加。因此认为,腺泡细胞内消化酶原与溶酶体水解酶相遇,是AP发生的早期事件。
NF-?资B激活与胰蛋白酶活化有关
大量实验已表明,所有腺泡细胞分泌的生理刺激物都是通过提升胞内Ca2+浓度而发挥作用的。体外实验观察到,用胆囊收缩素(CCK)孵育离体的腺泡细胞,胞内的Ca2+水平显著上升。AP时腺泡细胞内Ca2+浓度明显上升,用钙螯合剂可减轻胰腺炎程度。
从细胞生物学的角度看,腺泡细胞内钙失衡被认为是AP发生的始动因素。乙酰胆碱和蛙皮素可升高粗面内质网内的Ca2+水平,而CCK使溶酶体内的 Ca2+增加。胆酸可影响腺泡细胞内Ca2+水平,生理浓度的胆酸可通过腺泡细胞上的特殊膜转运蛋白进入胞内,抑制内质网上的Ca2+泵(Ca2+-ATP酶),使内质网内的Ca2+负荷增加。当各种病因导致胰管内压力升高或胰腺微循环障碍时,通过神经体液调节,腺泡细胞内Ca2+水平升高,一方面使含有溶酶体酶的细胞器质膜脆性增加,增加胞内溶酶体与酶原颗粒融合;另一方面使消化酶原与溶酶体水解酶进入高尔基器后,出现“分选”错误,在腺泡细胞内发生自我消化,并波及到胰腺间质。
此外,腺泡细胞内Ca2+水平升高还可活化核因子?资B(NF-?资B)。这是近年在AP发生机制研究上一个相当热门的话题。目前认为,NF-?资B的激活与胰蛋白酶活化有关,它不但启动局部炎症反应,也是全身炎症反应的扳机。但有趣的是,不同刺激物引起的NF-?资B活化,对腺泡细胞的命运影响差别甚大。如CCK引起的NF-?资B活化,可促进腺泡细胞的程序性死亡。肿瘤坏死因子α(TNFα)虽也可引起NF-?资B活化,但对腺泡细胞功能却没有明显影响。AP时,NF-?资B活化不仅限于胰腺,实际上累及多系统。转基因小鼠研究已表明,AP时肺和肝脏的NF-?资B活化增加,提示肝脏参与了AP引起的全身炎症反应综合征的调节。
新近研究观察到,胰蛋白酶激活的早期即有中性粒细胞浸润,而由中性粒细胞释放的辅酶Ⅱ氧化酶不断吸引中性粒细胞浸润和活化。浸润于组织中的中性粒细胞则通过呼吸爆发释放毒性内容物达到损伤组织,使炎症扩大,其在全身炎症反应综合征中的作用已较为明确。此外,环氧合酶2在实验性AP发生中起重要促进作用,当给予环氧合酶2抑制剂后或在环氧合酶2基因敲除动物的实验中,发现AP的炎症程度及伴随的肺损伤均明显减轻。